
Entrando em maiores detalhes …
A Atrofia Muscular Espinhal (SMA) é uma das principais causa de morte na criança. É uma doença autossómica recessiva, sendo a mais comum a seguir a Fibrose Cística, ocorre em aproximadamente 1 em 6.000 nascidos vivos.
No SMA, os cornos anteriores da medula espinal (primeiro neurónio motor) morrem, tendo por resultado o enfraquecimento progressivo dos músculos e finalmente, em alguns casos, a incapacidade de respirar e engolir.
O gene que é mutante no SMA tem sido clonado(a) , mas temos ainda pouca compreensão sobre como a proteína codificada é implicada no metabolismo do RNA de forma a permitir a sobrevivência dos neurónios motores. No artigo SMN Tudor domain structure and its interaction with the Sm proteins January 2001 Volume 8 Number 1 pp , Michael Sattler e os colegas apresentam a estrutura de um domínio desta proteína, e estes resultados são colocados no contexto da maquinaria celular pela notícia vista no artigo Tudor reign January 2001 Volume 8 Number 1 pp .
{kind=link}
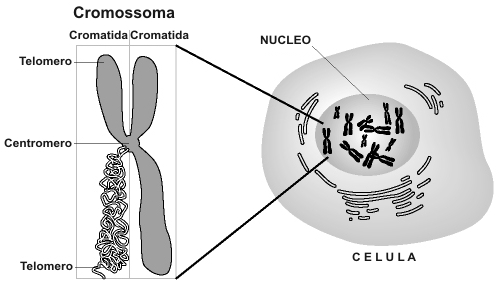
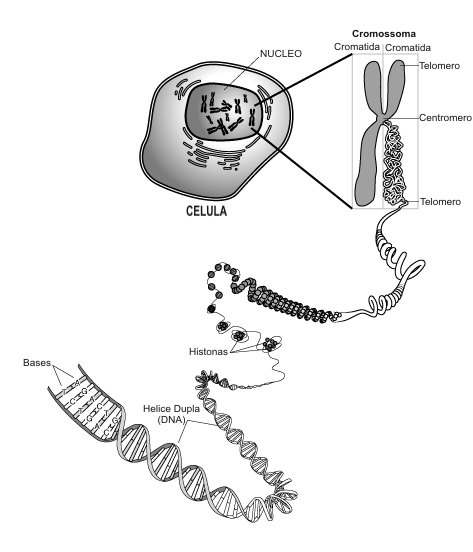
Há três tipos clínicos principais de SMA, com fenotipos(b) que variam desde os problemas musculares severos na infância (conduzindo à morte antes dos dois anos de idade) até simples fraqueza muscular no adulto. Em 1995, o gene telomérico SMN1 foi clonado no cromossoma 5(1) . Cerca de 95% dos casos de SMA são ligados ao gene SMN1; a maioria envolvem deleções(c) neste gene, mas, mutações pontuais podem também causar a doença. Pelo menos outros dois genes – SMN2 (que é quase idêntico ao gene SMN1) e o gene NAIP que codifica a proteína inibidora da apoptose(d) neuronal – afetam a severidade dos casos de SMA.
{kind=link}
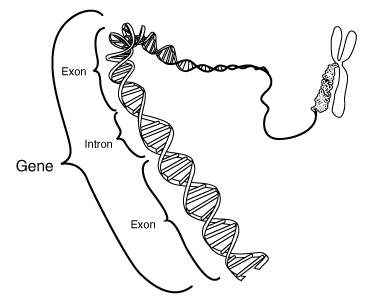
O gene SMN2 encontra-se perto do centrómero no cromossoma 5, por vezes em cópias múltiplas. Difere do SMN1 somente em algumas bases . Algumas mudanças nas bases afectam a emenda da transcrição do SMN2, tendo por resultado que o exon realçado salta(2) , o que produz uma proteína SMN2 truncada que não pode substituir inteiramente o SMN1. O Rato embora tenha o gene SMN1 não tem o gene SMN2 e Smn–/– embriões do rato morrem durante o implantação no útero. Entretanto, se o gene humano SMN2 estiver colocado no fundo genético Smn–/– do rato, os ratos sobrevivem e exibem um fenotipo SMA, apresentando fraqueza muscular e mudanças na medula espinal(3). O gene NAIP fica situado ao lado do gene SMN1 e esta suprimido frequentemente em conjunto com o gene SMN1 em pacientes com SMA. Uma perda da função de NAIP pode exagerar a severidade da doença permitindo uma apoptose neuronal aumentada.
{kind=link}
Diversas linhas da evidência implicam a totalidade da proteína de SMN no metabolismo do RNA (especificamente na emenda do RNA) (4) , (5) . Momentaneamente o gene SMN participa na montagem da partícula “pequena ribonucleoproteina nuclear” (snRNP) no citoplasma (provavelmente com sua interação com os componentes essenciais do snRNP chamados proteínas Sm) e pode também afetar a emenda mais directamente no núcleo (talvez ligando o RNA). Diversas mutações de SMN encontradas em pacientes de SMA cortam as interações SMN–Sm(6) , sugerindo que o papel de SMN no conjunto da partícula do snRNP é certamente relevante para à doença, embora a prova rigorosa desta hipótese seja requerida ainda. Estas observações são intrigantes porque emendar o RNA é uma função essencial em todas as pilhas humanas, e assim poderia esperar-se que tecidos adicionais fossem afetados em um indivíduo que abriga mutações de SMN. Isto poderia facilmente ser explicado se o gene SMN tivesse somente expressão nos neurónios motores, mas não é o caso. Assim, somos deixados perplexos com uma pergunta – como puderam as mutações no gene SMN conduzir somente à morte de neurónios motores e não a um fenotipo mais difundido?
Uma parte da dificuldade em compreender a especificidade de tais doenças neuronais, consiste no facto de sabermos muito pouco sobre a forma em como as várias populações neuronais se diferenciam – sobre os sinais moleculares que determinam que um dado neurónio se diferenciará em um neurónio do motor e não em um neurónio hipocampal. Por exemplo, para permitir o diferenciação e a sobrevivência do neurónio motor, pode ser que uma chave de transcrição encontrada somente nos neurónios motores e nos seus precursores deva ser emendada de uma maneira particular, e esta emenda poderia ser regulada pelo SMN. A pesquisa acerca das Stem Cells(e) humanas é crítica para descobrir tais especificidades.
(1) - Lefebvre, S. Pilha 80, 155-165 (1995). (2) - C.L., Hahnen, E., Androphy, E.J. & Wirth, B. Proc. Natl. Acad. Sci. EUA 96, 6307-6311 (1999). (3) - H.M. et al. Natureza Genet. 24, 66-70 (2000). (4) - Liu, Q., Fischer, U., Wang, F. & Dreyfuss, G. Pilha 90, 1013-1021 (1997). (5) - Pellizzoni, L., Kataoka, N., Charroux, B. & Dreyfuss, G. Pilha 95, 615-624 (1998). (6) - Pellizzoni, L., Charroux, B. & Dreyfuss, G. Biochemistry 96, 11167-11172 (1999). (a) - Clonagem Processo pelo qual são feitas copias de uma determinada região de DNA habitualmente um Gene. Quando um geneticista fala em clonagem ele não esta a pensar em copias genéticas de um organismo inteiro. (b) - Fenotipo Conjunto de características exteriores de um individuo que no seu conjunto ajudam a identificar de forma mais clara um quadro clínico. (c) - Delecção Zona de um gene sem informação. (d) - Apoptose Morte celular fisiológica, sendo o termo apoptose introduzido para se referir a este processo na década de 70. A supressão, aumento da expressão ou mutação dos diversos genes que atuam na apoptose parece estar associada a um grande número de doenças. Estas podem ser divididas em dois grandes grupos: Doenças onde há um aumento na sobrevivência das células associada a uma inibição da apoptose (câncer, doenças auto-imunes e infeções virais) Doenças onde ocorre um aumento na taxa de morte celular associada a uma maior activação do processo de apoptose (AIDS, doenças neurodegenerativas como Alzheimer, Parkinson, esclerose lateral amiotrófica, epilepsia, lesão cerebral no alcoolismo, isquemia cerebral e miocárdica, SMA). (e) - Stem Cells Também conhecidas como células pluripotentes, são as precursoras de todos os tipos celulares, encontram-se abundantemente no embrião.
Comentários recentes